-
Long-lived triplet charge-separated state in naphthalenediimide based donor–acceptor systems
A. Aster, C. Rumble, A.-B. Bornhof, H.-H. Huang, N. Sakai, T. olomek, S. Matile and E. Vauthey
Chemical Science, 12 (2021), p4908-4915


DOI:10.1039/D1SC00285F | unige:150871 | Abstract | Article HTML | Article PDF | Supporting Info
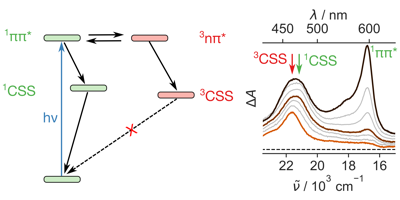
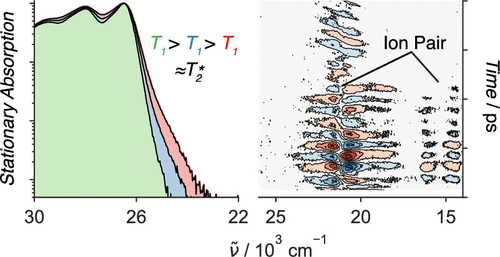
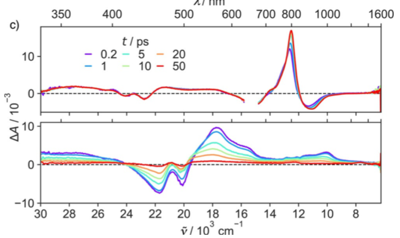
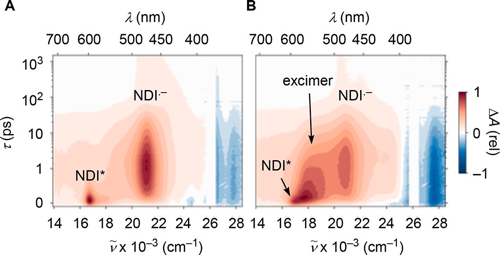
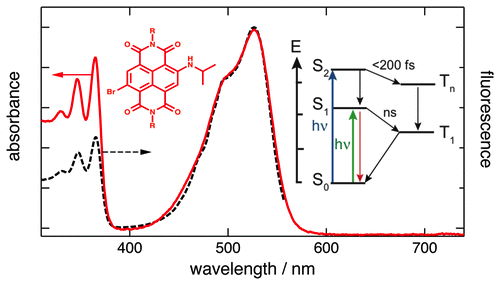
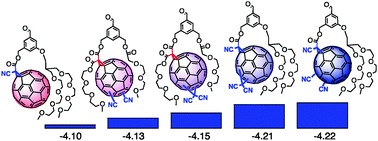
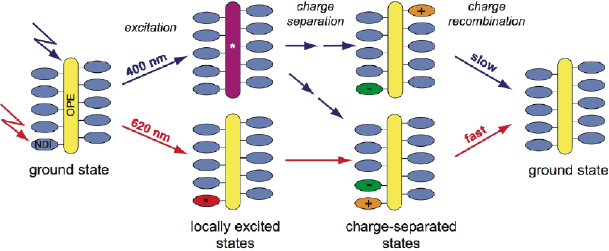
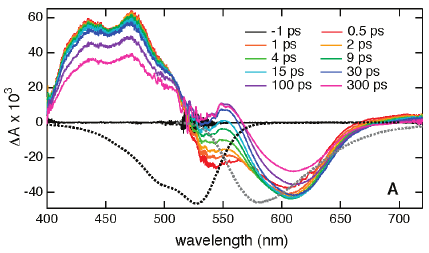
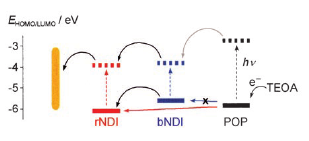
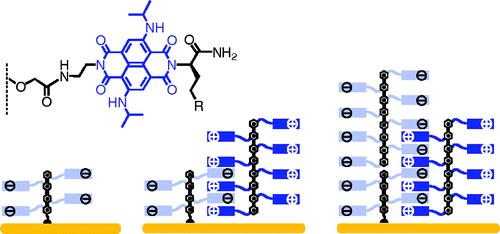
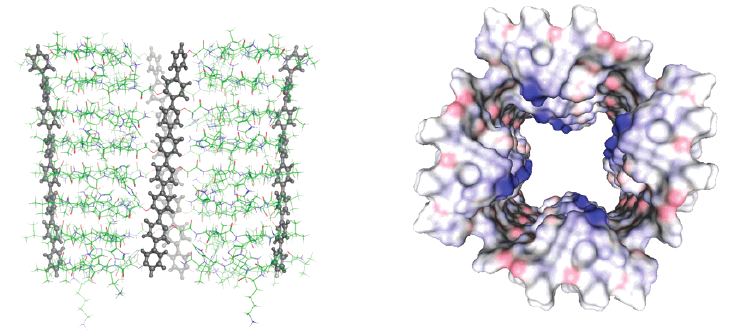
1,4,5,8-Naphthalenediimides (NDIs) are widely used motifs to design multichromophoric architectures due to their ease of functionalisation, their high oxidative power and the stability of their radical anion. The NDI building block can be incorporated in supramolecular systems by either core or imide functionalization. We report on the charge-transfer dynamics of a series of electron donorââ¬âacceptor dyads consisting of a NDI chromophore with one or two donors linked at the axial, imide position. Photo-population of the core-centred Ãâ¬Ã¢â¬âÃâ¬* state is followed by ultrafast electron transfer from the electron donor to the NDI. Due to a solvent dependent singletââ¬âtriplet equilibrium inherent to the NDI core, both singlet and triplet charge-separated states are populated. We demonstrate that long-lived charge separation in the triplet state can be achieved by controlling the mutual orientation of the donorââ¬âacceptor sub-units. By extending this study to a supramolecular NDI-based cage, we also show that the triplet charge-separation yield can be increased by tuning the environment.